All published articles of this journal are available on ScienceDirect.

Borrelia Genotyping in Lyme Disease
Abstract
Borrelia burgdorferi sensu lato is the causative agent of Lyme borreliosis, multisystem disorder characterized by a wide spectrum of clinical manifestations. Different borrelia species can lead to distinct clinical presentations, but some species were associated with defined clinical manifestation like Borrelia afzelii with skin manifestations, Borrelia garinii with central nervous system disorders and Borrelia burgdorferi sensu stricto with Lyme arthritis. Ixodes ticks represent the main vectors of B. burgdorferi sensu lato; wild animals, lizards and birds are the natural reservoir of borrelia. Genotyping of borrelia strains is of great importance for epidemiological, clinical, and evolutionary studies. Numerous methods are available for the genotyping of B. burgdorferi sensu lato based either on whole genome or PCR based typing. Typing methods differ in their approach and target, many of them were implemented more or less successfully for diagnostic purposes.
INTRODUCTION
Borrelia burgdorferi sensu lato is the causative agent of Lyme borreliosis, multisystem disorder characterized by a wide range of clinical symptoms. Usually, the initial clinical manifestation of borrelial infection is localized skin disorders, erythema migrans, from which borrelia may disseminate to various organs causing early disseminated (multiple erythema migrans, Lyme neuroborreliosis, Lyme carditis, borrelial lymphocytoma) or persistent infection (acrodermatitis chronica atrophicans, chronic Lyme arthritis, late neurological manifestations) [1]. Different borrelia species were reported to cause distinct clinical manifestations, whereas some species were associated with defined clinical manifestation like B. afzelii with skin manifestations, B. garinii with central nervous system disorders and Borrelia burgdorferi sensu stricto with Lyme arthritis [1].
The Lyme borreliosis complex of spirochetes includes at least 20 named genospecies [2, 3]. Five of these species, Borrelia afzelii, Borrelia garinii, Borrelia bavariensis, Borrelia burgdorferi sensu stricto and Borrelia spielmanii have been described as the causative agents of Lyme borreliosis in humans [1]. Other borrelia species (Borrelia lusitaniae, Borrelia bissettii, Borrelia valaisiana) were rarely or never isolated from humans and their pathogenicity remains unclear. The aim of many research studies is to assess the relationship of particular borrelia species and its organotropism based on molecular level.
Natural reservoir of borrelia are wild animals, lizards and birds. Specific rodents were identified as the principal reservoir of B. burgdorferi sensu lato in Europe like wood mouse (Apodemus sylvaticus), yellow-necked mouse (Apodemus flavicollis) and bank vole (Clethrionomys glareous) [4, 5]. Several other mammals, were also described as competent reservoirs like dormice, hedgehogs, rats, squirrels, hares, other [6]. Strains of particular borrelia species are associated with particular reservoir hosts that was explained by borrelial susceptibility/resistance to host’s complement system. Irrespective to the host, excluding human, borrelial infection is life lasting and generally not harmful for the host. Today, many research works are based on molecular and genetic analysis of borrelial association with its host.
Ixodes ticks represent the main vectors of B. burgdorferi sensu lato, in Europe mainly Ixodes ricinus, in Asia Ixodes persulcatus, in northeastern and upper midwestern USA Ixodes scapularis and in western USA Ixodes pacificus [1]. Different competent tick vectors are able to acquire, maintain, and transmit borrelia from one host to another, including humans. Although borrelia is fastidious for cultivation it is able to survive large temperature range and unfavorable conditions as is exposed in ticks during the ticks’ life cycles through winters and summers. Molecular basis of this livelihood is still unclear [7].
B. burgdorferi sensu lato is a typical spirochete, motile, host associated and demanding for cultivation. Very unusual is borrelial genome that consists linear chromosome (of approximately 910 kbp) and numerous linear and circular plasmids that can contain over 600 kbp of DNA [8]. All members of the Lyme borreliosis spirochetes that have been already analyzed carry linear chromosome similar in size to the chromosome of the strain B31 [8]. The linear replicons have covalently closed telomeres [9]. Majority of housekeeping genes are localized on the chromosome, whereas genes encoding different lipoproteins that are expressed on the bacterial outer membrane are positioned on the plasmids. Among borrelial strains, plasmid content differs and presents borrelial make up in nature. Some parts of borrelial genome are specific for borrelia like chromosome carrying a single gene encoding 16S rRNA (rrs) separated from a tandemly repeated pair of 23S (rrlA and rrlB) and 5S rRNA (rrfA and rrfB) genes; this unique rRNA gene organization presents target for borrelia molecular analysis [10].
Genotyping of borrelia strains is of great importance for epidemiological, clinical, and evolutionary studies. Numerous methods are available, all based either on whole genome typing or PCR based typing. Typing methods differ in their approach and target; many of them were implemented more or less successfully for diagnostic purposes. Herein we present some typing methods.
Whole genome based genotyping presents robust methods for borrelia characterization; it includes species identification, plasmid profile analysis and whole genome sequencing.
WHOLE GENOME BASED RESTRICTION
Large restriction fragment pattern (LRFP) requires growing borrelia culture; it is based on whole genome restriction analysis [11]. Different restriction enzymes can be employed for restriction of genomic DNA, MluI, ApaI, KspI, SmaI, and XhoI. MluI based restriction is most commonly used regarding to its potential of borrelia species determination [12]. Restricted genomic DNA is separated using pulse-field gel electrophoresis, where large DNA molecules are separated due to periodical changes in electric field. Restriction based on MluI enables identification of borrelia species and delineation of subgroups within the species. Some MluI-based LRFP are presented in (Table 1).
MluI-LRFP of B. afzelii isolates shows quite homogeneous restriction pattern; the majority of isolates belong to B. afzelii Mla1 subgroup (>99 %), and minority to B. afzelii Mla2, Mla3 and Mla4 [11].
On the other hand, B. garinii and B. burgdorferi sensu stricto isolated strains have very heterogeneous restriction patterns and are divided into 7 (Mlg1 – 7) and 15 (Mlb1 – 15) subgroups, respectively [11], as presented in (Table 1). Some of these subgroups were more frequently associated either with geographic region, reservoir host or clinical manifestation. Due to the demanding approach there are just a few studies regarding the subject [11].
B. valaisiana, B. lusitaniae and B. spielmanii have quite homogenous restriction patterns, only two subgroups were identified in each species, but not many strains were analyzed [11].
PLASMID PROFILE ANALYSIS
Determination of plasmid profile requires growing borrelial culture. Genomic borrelial DNA can be isolated by gel-insert method as described previously [11, 13, 14]. Briefly, bacterial cells with density of 109/ml are embedded in agarose blocks, lysed with lysozyme and digested with proteinase K [11, 13, 14]. For separation of chromosomal and plasmid DNA pulse-field gel electrophoresis (PFGE) with ramping time of 0.9–3 s and run time of 37 h is used [13, 15, 16]. The relative molecular size of particular linear plasmid should be calculated regarding to appropriate pulse marker. Using PFGE only linear plasmids can be separated that is disadvantage of the methods regarding to borrelial circular plasmids. Linear plasmid profiles of particular strain are established based on the number of plasmids per cell and molecular mass of the plasmids [17].
Among borrelia strains, number of linear plasmid vary as well as their size [18]. The presence of multiple plasmids of same molecular mass that can be identified only by PCR was also confirmed. Additionally, some plasmids can be lost during pro-long cultivation [19]. Low-copy number of particular plasmid that is below PFGE sensitivity, makes plasmid invisible and determination of plasmid profile more difficult [18]. Some publications report that every strain possesses one large and numerous small plasmids while the others report strains with multiple large plasmids and/or plasmid dimers, both unusual for spirochets [20, 21].
NEXT GENERATION SEQUENCING
Next generation sequencing enables generating whole-genome sequences for numerous bacterial isolates on a single sequencing instrument in less than one day. Major disadvantage of the methods represents, besides its expenses, a lack of user-friendly data analysis and interpretation tools [22]. Several platforms can be used for next generation sequencing like 454 (FLX Titanium), Illumina (HiSeq, MiSeq, GA), SOLiD (4, 5500), Helicos, Ion Torrent, PacBio and Starlight. Up to now, complete or partial whole genome sequences of 42 B. burgdorferi sensu stricto, 9 B. afzelii and 40 B. garinii isolates are available (NCBI Genome; http://www.ncbi.nlm.nih.gov/genome). Next generation sequencing was also used in research of Troy et al. to gain insight in B. burgdorferi sensu stricto dissemination during infection [23]. They developed massively parallel sequencing combined with transposon mutagenesis to study B. burgdorferi sensu stricto pathogenesis. The results demonstrated that B. burgdorferi sensu stricto is a highly successful pathogen that, after surviving a population bottleneck at the site of inoculation, is able to spread throughout a mammalian host relatively unimpeded in order to establish long-term infection [23].
PCR-BASED TYPING
PCR-based restriction fragment length polymorphism (RFLP) analysis utilizes PCR amplification of either the rrs-rrlA (16S-23S) or the rrfA-rrlB (5S-23S rRNA) spacer followed by restriction endonuclease digestion of the PCR product and separation of the fragments by gel electrophoresis [24, 25]. The technique can be employed for typing of cultivated spirochetes (one step PCR) or uncultivated spirochetes from clinical or tick samples (nested PCR).
Amplification of the rrfA-rrlB spacer results in amplicon sizing 225 to 266 bp. Two restriction enzymes, MseI and DraI, are most often used for RFLP. Eight distinct species of B. burfdorferi sensu lato can be differentiated using MseI. Resolution of restriction digests can be visualized using 16% acrylamide–0.8% bisacrylamide gels [24]. This approach is widely used for strain identification and also for direct identification from clinical specimens, ticks or reservoir host [2, 26-31]. Fig. (1) shows MseI restriction of rrfA-rrlB (5S-23S rRNA) spacer of different borrelia.
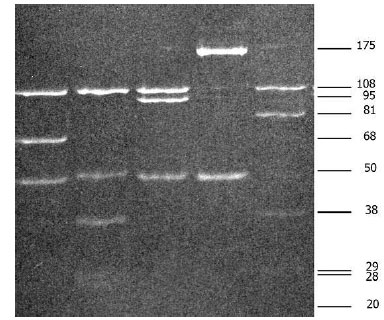
MseI restriction of amplified rrfA-rrlB (5S-23S rRNA) spacer of Borrelia burgdorferi sensu lato strains: Lane 1: Borrelia afzelii, Lane 2: Borrelia burgdorferi sensu stricto, Lane 3: Borrelia garinii, Lane 4: Borrelia valaisiana, Lane 5: Borrelia lusitaniae.
The rRNA spacer rrs-rrlA is amplified using a nested PCR, resulting in 941-bp amplicon. RFLP analysis using HinfI or MseI differentiates B. burgdorferi sensu stricto strains into three ribosomal spacer types, RST1, RST2, and RST3 [32]. According to literature B. burgdorferi sensu stricto RST types correlate with pathogenic potential [32-36]. A significantly higher percentage of patients with Lyme borreliosis who were infected with RST1 strains had more often positive blood culture results, multiple erythema migrans, and more severe symptoms in comparison to patients infected with B. burgdorferi sensu stricto RST2 or RST3 isolates [35].
OUTER SURFACE PROTEIN C (OSPC) ANALYSIS
Gene for OspC is located at single-copy circular plasmid cp26, known to be essential for in vitro growth [9]. OspC plays critical role in transmission of borrelia from tick to vertrebrate, also for borrelial infectivity in vertebrate [37-39]. OspC is one of the most immunodominant antigens in the humoral IgM immune response [40].
Genotyping employs amplification and sequencing of an approximately 600 bp region of the OspC gene [41]. OspC typing divides B. burgdorferi sensu lato strains into 21 genetically distinct types. A correlation between ribosomal spacer types and OspC genotypes has been established for B. burgdorferi sensu stricto: RST1 corresponds to OspC genotypes A and B; RST2 to OspC types F, H, K, and N; and RST3 to the remaining 10 OspC types, including D, E, G, and I [36, 42-44].
Comparing RST and OspC genotyping of isolated strains revealed that RST can miss differences within groups while OspC genotyping may lead to small groups; by both typing systems, more information with clinical correlations can be obtained. OspC as well as other outer surface proteins are variable and frequently used in population studies within the species [45].
Borrelia burgdorferi sensu lato subgroups determination using MluI large restriction fragment pattern (MluI-LRFP): Mla – subgroups of Borrelia afzelii, Mlg – subgroups of Borrelia garinii, Mlb– subgroups of Borrelia burgdorferi sensu stricto, Mls– subgroups of Borrelia spielmanii, Mlv– subgroups of Borrelia valaisiana, and Mll– subgroups of Borrelia lusitaniae.
 |
REAL-TIME PCR AND MELTING TEMPERATURE ANALYSIS
Real-time PCR combined with melting temperature determination of amplified target DNA facilitates borrelia identification. Melting temperature is highly consistent for DNA fragment, defined by the nucleotide sequence, its length, and GC content [46, 47]. Appropriately created protocol can be used to distinguish bacteria on species level. For identification of borrelia species several borrelial genes (hbb, p66, recA, ospA, etc.) were employed [48-50].
Identification based on differences in hbb gene enables distinguishing majority of Lyme disease borrelia species although it fails to discriminate B. spielmanii from B. valaisiana [51]. Fig. (2) shows real-time PCR combined with melting temperature determination of hbb gene.
FLAGELLIN BASED TYPING
The flagellin gene, located on chromosome is highly conserved and in some parts quite diverse among different B. burgdorferi sensu lato species. Because of its’ easy access, it is generally used for diagnostic purpose while its’ diversity can be used for borrelia species identification [52, 53].
Jaulhac et al. described oligonucleotide typing method with PCR fragments from the flagellin gene of B. burgdorferi sensu lato, which is able to differentiate seven different borrelia species, B. garinii, B. afzelii, B. burgdorferi sensu stricto, B. japonica, B. andersoni, B. valaisiana, B. bissettii [54]. Method is technically demanding and is used mainly for borrelial confirmation in clinical samples.
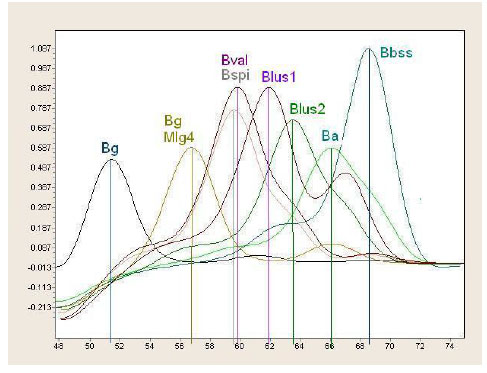
Real-time PCR combined with melting temperature determination of hbb gene (Bg - Borrelia garinii; Bval - Borrelia valaisiana; Bspi - Borrelia spilmanii; Blus - Borrelia lusitaniae; Ba – Borrelia afzelii; Bbss - Borrelia burgdorferi sensu stricto).
MULTILOCUS SEQUENCE TYPING
Multilocus sequence typing (MLST), as defined by Urwin and Maiden, is based on amplification, sequencing, and bioinformatic analysis of internal fragments of housekeeping genes that evolve slowly and are located throughout the genome to avoid bias [55]. MLST represents a molecular typing tool that can be used for the population studies, phylogenetic analysis, epidemiological monitoring and evolutionary studies [55, 56].
For borrelia spirochets, eight housekeeping loci have been chosen, clpA (Clp protease subunit A), clpX (Clp protease subunit X), nifS (Aminotransferase), pepX (Dipeptidylaminopeptidase), pyrG (CTP synthase), recG (DNA recombinase), rplB (50 S ribosomal protein) and uvrA (Exonuclease ABC) [57]. The Borrelia MLST scheme is available through the MLST network (http://www.mlst.net/). This method delineated Borrelia bavariensis from other B. garini strains regarding to some epidemiological data of the strain (birds were determined as reservoir of B. garini while small mammals of B. bavariensis) [2]. Generally, MLST was proposed as alternative method for borrelial classification, phylogeny and ecological studies of spirochets.
CONCLUSION
Numerous methods based either on whole genome or PCR based typing are available for the genotyping of B. burgdorferi sensu lato. Typing methods differ in their approach and target, many of them were implemented more or less successfully for diagnostic purposes. The aim of all molecular genotyping is to assess relationship between borrelia and its reservoir hosts and vectors as well as to clarify molecular background of pathogenicity.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGeMENTS
Declared none.

