All published articles of this journal are available on ScienceDirect.

Intrafamilial Diversity of Clinical Severity in Dominant Dystrophic Epidermolysis Bullosa: Case Series of Three Generations
Authors Info & Affiliations
Abstract
Background:
Epidermolysis bullosa, a group of inheritable blistering diseases with considerable clinical and genetic diversity, is divided into distinct subtypes depending on the level of tissue separation in the dermal–epidermal basement membrane zone. The dystrophic form of epidermolysis bullosa (DEB) is characterized by tense blisters and erosions which heals with extensive scarring. The fact that DEB can be inherited in either autosomal dominant (DDEB) or autosomal recessive (RDEB) pattern adds to its clinical diversity. The cause of marked clinical diversity in mild to severe DDEB is still unidentified.
Main Observation:
We report an intrafamilial diversity of clinical severity in dominant dystrophic epidermolysis bullosa (DDEB) cases within three generations.
Conclusion:
We emphasize the variety of clinical severity in DDEB cases within three generations which might be caused by unknown gene modifiers and environmental factors.
1. INTRODUCTION
Epidermolysis Bullosa (EB) is a group of inherited mechanobullous blistering disorders characterized by fragile skin and mucous membrane. The cause of EB is mutation of various structural proteins in the skin resulting in the various levels of tissue separation within the dermal-epidermal basement membrane zone [1, 2]. There have been several advances in the classification of EB since it was originally proposed in 1991 [3]. Dystrophic EB (DEB) can be inherited in either autosomal dominant (DDEB) or autosomal recessive (RDEB) pattern. The phenotypic spectrum observed in families with DEB result from mutations of COL7A1 gene at the mRNA and protein level in combination with environmental factors [4]. The cause of clinical diversity of mild to severe DDEB is still unknown, but it is implied that there may be substantial effects from another genetic and/or non-genetic disease modifiers [5]. To the extent of the author’s knowledge, this is the first report of diverse clinical severity of DDEB in three generations.
2. CASE SERIES
We report five DDEB cases in one family with diverse clinical severity. The pedigree of this family is displayed in Fig. (1).
2.1. Patient 1
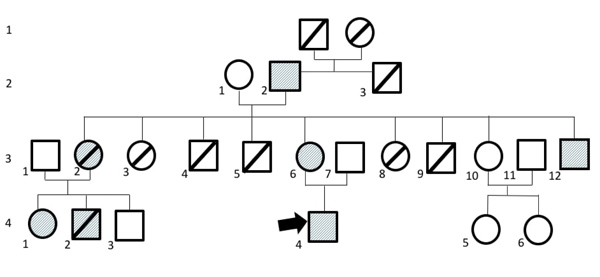
A 57-year-old male, the grandfather of the proband described below, had a history of hemorrhagic blisters and erosive lesions on all over the body since birth. Some blisters and erosive lesions progressed into ulcers with crusted surface on upper and lower extremities. The lesions often recurred following friction or trauma. Almost all lesions healed with scar formation. There were dystrophic fingers and toenails (Fig. 2). There was no abnormality on the hair and teeth. He recalled that his parents had no clinical features of DDEB. Body Surface Area (BSA) involvement was 36%. Epidermolysis Bullosa Disease Activity and Scarring Index (EBDASI) score was 39.
Notes: Affected individuals: stripe; death: slash
Patient 1 (2.2) had extensive skin blistering and dystrophic nails. The lesions healed with scar formation.
Patient 2 (3.6) & patient 3 (3.12) had extensive skin blistering, erosions, and dystrophic nails.
Patient 4 (4.1) had skin blisters, erosions, and dystrophic nails with mucous involvement. The lesions healed with scar formation.
Patient 5 (4.4) had minor blistering on area with friction and erosions, with nail involvement. Lesions healed with minor scar formation.
2.2. Patient 2
A 32-year-old female, the mother of the proband, had a history of hemorrhagic blisters, bullae, and few erosive lesions on upper and lower extremities that extended throughout the body since birth. The lesions often recurred following friction or trauma. Almost all lesions healed with scar formation. There were some dystrophic changes in the fingernails and toenails (Fig. 2). There was no abnormality on the mucous membranes, hair, and teeth. Her father, brother, nephew, and son had similar clinical features, indicating autosomal dominant inheritance. BSA involvement was 63%. EBDASI score was 20.
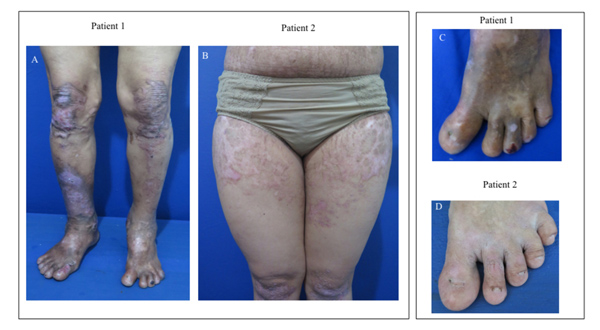
2.3. Patient 3
A 22-year-old male, the uncle of the proband had persistent atrophic scars and hyperpigmentation. He had a history of hemorrhagic blisters and erosions with crusted surface on upper and lower extremities that extended throughout the body since birth. The lesions often recurred due to friction or trauma. Almost all lesions healed with scar formation (Fig. 3). There were changes in his fingers and toenails without the involvement of mucous membranes, hair, and teeth. His father and sister had similar clinical features of DDEB. BSA involvement was 72%. EBDASI score was 23.
2.4. Patient 4
A 15-year-old girl, the cousin of the proband, had a history of hemorrhagic blisters, bullae, and erosive lesions on upper and lower extremities that extended throughout the body since birth. The lesions often recurred following friction or trauma. Almost all lesions healed with scar formation. There was no abnormality on the mucous membranes, hair, and teeth. Dystrophic nails were observed on the hands and feet (Fig. 4). She informed that her mother, aunt, and paternal grandfather had similar clinical features of DDEB. BSA involvement was 45%. EBDASI score was 24.
2.5. Patient 5
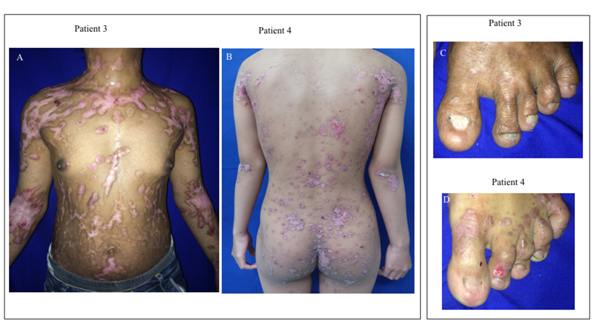
A 2-year-old boy, the proband, had a history of blisters, bullae, and few erosive lesions restricted on both legs and hands since birth with different clinical features compared with the other family members. At the age of 2 weeks, he had minor blistering on the feet with neither scar formation, nor milia and nail involvement (Fig. 4A-B). At the age of 1 year, the lesions healed with scaring and milia developed on dorsal hands and back and followed with nail dystrophy (Fig. 4C-D). There was no abnormality in the mucous membranes, hair, and teeth. BSA involvement was 23%. EBDASI score was 15.
We performed genetic testing with Miseq Illumina system®. Genetic testing of all patients revealed mutations of c.6218G>A:p.(Gly2073Asp) and c.5945G>T:p.(Gly1982Val) in COL7A1 which confirmed the diagnosis of DDEB.
3. DICUSSION
Dystrophic EB, manifesting as blister formation beneath the lamina densa within the papillary dermis, is divided into two forms based on the inheritance pattern: autosomal dominant and autosomal recessive form [6]. In regard to the genetic background, both DDEB and RDEB are caused by mutations in the COL7A1 gene which encodes type VII collagen [7]. DDEB is the second most common major type of EB following epidermolysis bullosa simplex (EBS), supposedly due to its autosomal dominant pattern. In DEB, different phenotypes are caused by heterozygous, compound heterozygous, or homozygous mutations in COL7A1 gene [8].
The dystrophic form of EB is characterized by skin fragility which heals with extensive scarring. [2] The scarring tendency indicates that blister formation in DEB occurs below the lamina densa of the cutaneous basement membrane, eliciting a mesenchymal wound-healing response in the dermis. In addition to cutaneous blisters and erosions, the gastrointestinal tract, particularly the esophagus, is also affected by blistering and scarring. [4] The extensive scarring on the hands and feet is often associated with the development of aggressive, rapidly metastasizing squamous-cell carcinomas, which can lead to early death [2].
However, the spectrum of clinical severity in DEB is highly variable, with milder form presenting a limited tendency of blistering or nail involvement. [4] DEB can be inherited in either autosomal dominant or an autosomal recessive pattern (DDEB and RDEB, respectively). The clinical severity is highly variable, demonstrated as a spectrum of phenotypic manifestations [2].
The phenotype of DDEB consists of blisters, milia, scarring, and dystrophic nails. In general, it does not lead to an increased risk of dental caries, growth retardation, cutaneous malignancies, anemia, pseudosyndactyly, or death. Mostly, DDEB have a milder phenotype in contrast to RDEB, which manifests as excessive blistering, atrophic scarring, and a lot of other cutaneous and extracutaneous findings, pseudosyndactyly, cutaneous malignancies, as well as increased risk of death [9].
The EBDASI has demonstrated excellent reliability, validity, and feasibility which quantifies the overall severity of the involved skin, scalp, mucous membranes, nails, and other epithelialized surfaces based on the disease activity and damage. This scoring is suitable for all EB subtypes and ages. The EBDASI is more reliable in distinguishing minor severity differences compared to The Birmingham EB Severity, particularly for mild to moderately severe EB [10].
We report five DDEB cases in a family because the clinical manifestations revealed tense bullae, erosions, and nail involvement. Lesions healed with scarring, milia formation, and nail dystrophy, but there was no mucosal and organ involvement. Therefore, we considered DDEB pattern inherited in this family. We calculated the total activity and damage score based on EBDASI resulting in different clinical severity in all patients.
The predominant mutation in the DDEB is a missense mutation resulting in glycine substitution within the triple helical domain of type VII collagen [11]. Most of these mutations tend to cluster within exons 73–75, suggesting that the location of a glycine in every third position in this segment of type VII collagen is critical for the stability of the triple helix [4]. A glycine substitution at the same position that results in a different amino acid residue can also give rise to variable phenotypic expression [6].
The proband (patient 5) had the clinical manifestation similar to EBS marked by healed lesions without scarring at the age of two weeks. Furthermore, the lesions were changing and healed with scarring and nail involvement as the age increases. We hypothesized that the youngest member had a different mutation in the form of glycine substitution in COL7A1 combined with environmental factors which resulted in different clinical severity from other family members. The genetic testing revealed the same mutation, c.6218G > A:p. (Gly2073Asp) and c.5945G > T:p.(Gly1982Val) in COL7A1. Because scarring and its sequelae become more apparent with increasing age, some of the DEB manifestation may not be found in infants or children [12]. Therefore, we can explain the variation in clinical manifestations.
Similar cases have been reported by Shimizu et al. [8]. They reported a 10-year-old girl who was born in September 1988 as the second child of nonconsanguineous healthy parents of Japanese ethnic origin. She was diagnosed with DEB. At birth, she presented with blisters on the entire skin and mucous membranes. Although the severity of the skin condition had slightly decreased, the blister formation and nail deformities remained. At the age of 10 years, she experienced large scarring, ulcerated plaques, loss of all nails, and left toes deformity. Kasi et al. [13] showed a three-generation family with congenital central hypoventilation syndrome in four family members, suffering from a novel PHOX2B non-polyalanine repeat mutation (NPARM) with variable phenotypes which were inherited in autosomal dominant pattern. One possible cause for this phenotypic variability is unknown gene modifiers, while another possible underlying mechanism was environmental factors [13]. Early age cannot predict the ultimate phenotype [14]. Wide phenotypic variety is also observed across the different sub-types of DEB, which are demonstrated as multiple mutations resulting in a similar phenotype. Also, the same mutations show variable clinical features, in addition to considerable inter and intra-familial variations [14]. In our case, the family demonstrates the same mutation in COL7A1; however, they show phenotypic variability. The clinical manifestation might be not fully developed, particularly in patient 5. Considering that the Kasi et al. study’s case and our case have similarities in the autosomal dominant inheritance pattern, therefore the gene modifiers and environmental factor could be considered as a possible underlying mechanism for phenotypic variability.
CONCLUSION
Phenotypic variability could occur in one family with autosomal dominant pattern of DEB due to a combination of unknown gene modifiers and environmental factors. Accurate genetic counselling is essential for the identification of DDEB in the other family members and the education regarding the possibility of disease inheritance to the next generation. The use of genetic testing is important to confirm EB subtype as clinical assessments may be inaccurate, particularly in younger patients who do not demonstrate all clinical features of the EB subtypes.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the Health Research Ethic Committee of Dr. Hasan Sadikin General Hospital, Bandung, Indonesia.
HUMAN AND ANIMAL RIGHTS
No Animals were used in this research. All human research procedures followed were in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
Informed consent was obtained from all the patients when they were enrolled.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors are grateful to Universitas Padjadjaran, Bandung, Indonesia for financial support. (Grant number 872/UN6.3.1/LT/2017). The authors are also thankful to the authority of the Department of Dermatology and Venereology, Universitas Padjadjaran/Dr. Hasan Sadikin Hospital Bandung for affording necessary facilities. We thank Professor Mark Koh Jean Aan, MD and Tan Ene Choo, MD for their aid in conducting genetic testing for our patient.

