All published articles of this journal are available on ScienceDirect.

Lessons From Epithelialization: The Reason Behind Moist Wound Environment
Abstract
Wound healing consists of multiple structured mechanism and is influenced by various factors. Epithelialization is one of the major aspect in wound healing and inhibition of this mechanism will greatly impair wound healing. Epithelialization is a process where epithelial cells migrate upwards and repair the wounded area. This process is the most essential part in wound healing and occurs in proliferative phase of wound healing. Skin stem cells which reside in several locations of epidermis contribute in the re-epithelialization when the skin is damaged. Epithelialization process is activated by inflammatory signal and then keratinocyte migrate, differentiate and stratify to close the defect in the skin. Several theories of epithelialization model in wound healing have been proposed for decades and have shown the mechanism of epidermal cell migration during epithelialization even though the exact mechanism is still controversial. This process is known to be influenced by the wound environment where moist wound environment is preferred rather than dry wound environment. In dry wound environment, epithelialization is known to be inhibited because of scab or crust which is formed from dehydrated and dead cells. Moist wound environment enhances the epithelialization process by easier migration of epidermal cells, faster epithelialization, and prolonged presence of proteinases and growth factors. This article focuses on the epithelialization process in wound healing, epithelialization models, effects of wound environment on epithelialization and epithelialization as the basis for products that enhance wound healing.
1. INTRODUCTION
Skin epidermis consists of multi-layered stratified epithelium and is renewed over time. Skin homeostasis is maintained by proliferation of basal cell and differentiation of suprabasal cell and influenced by reciprocal signalling [1, 2].
Wound healing mechanics are complex which are influenced by various mechanism involving the coordinated interaction of blood cells, various enzymes, growth factors, and extracellular matrix. The different phases of wound healing can be categorized into inflammatory, proliferative and maturational phase. During the first phase i.e. the inflammatory phase, a blood clot forms and immune cells are concentrated to the wound site. Then, during the proliferative phase, keratinocytes from epidermis and dermis start proliferating and migrate into the wound bed. Lastly, in maturational phase, restructuration of the extracellular matrix occurs in the wound bed [1, 3].
Keratinocytes which migrate to the wound bed come from the adjacent epidermis, outer root sheath, and sweat duct stem cells. This process is dependent upon actin and myosin filament systems and the proliferative potential of these cells is inhibited while migrating [4-6].
Keratinocytes migration starts from the wound edges soon after wounding and inflammatory cells are recruited to the wound site by various chemotactic signalling. The trigger for keratinocyte motility post-wounding is probably because of the lost contact with the adjacent keratinocytes and basement membrane laminin- 5 reduction, which is mirrored by a decrease in integrin a6b4.
Migrating keratinocytes spreads under the scab debris and sometimes it might burrow a path under the hard scab and underlying granulation tissue to provide a new epithelial coverage until the two epithelial tongues meet in the centre of the wound. The direction of migration is controlled by the orientation of extracellular matrix macromolecules and other chemotactic factors that are present in the wound bed [5-7].
During the progression of this process, fibroblastic cells are concentrated to the wound bed and then secrete collagenous extracellular matrix. This process continuously replaces the fibrin eschar. Around the wound edges, a single layer of cells initially forms over the defect and there’s also a marked increase of epithelial cell mitotic activity. The endpoint of migration is when the advancing epithelial cells meet. Then, the basement membrane starts to form. After the epithelium reaches maturity, shedding and dispatching of the fibrous clot from the underlying epithelium occurs [5-7].
2. SKIN STEM CELL IN WOUND HEALING
Skin stem cell is known to reside in several locations as shown in Fig. (1). Epidermal stem cell is located in the interfollicular basal cell. Bulge stem cell is located in the hair follicle under arrector pili muscle insertion. Sebaceous gland stem cell is located in the base of sebaceous gland. During self renewal, these stem cells act independently. When epidermis is damaged, these stem cells can produce new cells from different kind of stem cells. This process occurs by reducing N tip from Left1 to inhibit β-cathenin signalling which induces trans-differentiation of hair follicle into interfollicular epidermis and sebocyte. Bulge stem cell can move into epidermis when epidermis is injured. Normally, this stem cell only acts in anagen, telogen and catagen hair cycle. This happens because this stem cell is multipotent and during its development there are some interactions between skin epithelial cell and mesenchymal which guide the cell differentiation into hair follicle, epidermis or sebaceous gland. Other than those three types of stem cell, there are also isthmus stem cell which are located in the central isthmus and junctional zone epidermis, mesenchymal stem cell in skin mesenchymal, melanocyte stem cell under the bulge stem cell, neural progenitor stem cell, and hematopoietic stem cell [8-10].
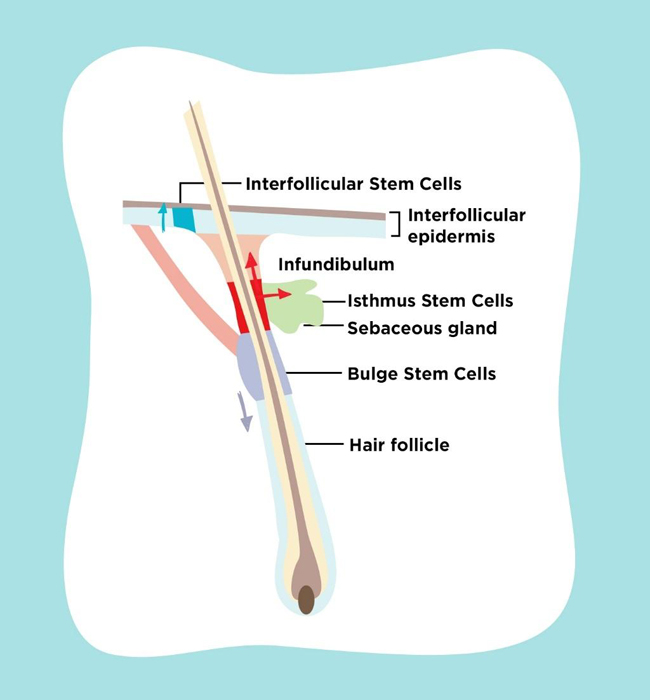
Under normal homeostatic conditions, each epidermal stem cell niche acts unipotently and replenishes its own respective tissue compartment. After injury to the skin, stem cells from the hair follicle and interfollicular epidermis contribute towards re-epithelialization of wounds. Previous research found that both interfollicular epidermis and hair follicle stem cells participate in wound healing, but hair follicle stem cells are not necessary for maintaining the interfollicular epidermis. It is also known that cells in the pilosebaceous unit become activated when skin is injured thus contributing towards interfollicular epidermis repair [11-16].
3. KERATINOCYTES PROFILES IN EPITHELIALIZATION
Epidermal keratinocytes are known to contain holoclones, paraclones, and meroclones according to their proliferative potential. Holoclones have the greatest proliferative potential and self-renewing abilities and can give rise to both meroclones and paraclones. Meroclones contain cells with various growth potential and can give rise to paraclones and meroclones when re-seeded. In contrast, paraclones only possess a short replicative life span [17-21].
Keratins play an important role in several processes such as transcription regulation, adhesion, migration, proliferation, epithelial polarity, angiogenesis, protein catabolism, and inflammatory regulation. Keratin intermediate filament proteins are epithelial cell cytoskeletal components that provide mechanical stability and protection from cell stress. These proteins exist as polymeric filaments by pairing of type I and type II keratin proteins, quickly respond to their cellular environment, and can be modulated during cell stress. Based on the histopathological studies, proliferative keratinocytes infrastructure in the basal layer are composed of K5 and K14. The cells move toward the surface traversing layers known as the spinous layer, granular layer, and stratum corneum. While these keratinocytes ascend and differentiate, it can be found that the synthesis of K5 and K14 in the basal layers switches to K1 and K10 in the suprabasal layers. Both basal cells attached to the basement membrane (hemidesmosomes and focal adhesions) and suprabasal cells attachedto their neighboring cells (desmosomes) have to be detached to allow keratinocyte migration in the re-epithelialization model. Further functional studies are still needed in the future [5, 22, 23].
4. EPITHELIALIZATION
Epithelialization is a process of covering defect on the epithelial surface during the proliferative phase that occurs during the hours after injury. In this process, keratinocytes renew continuously and migrate upward from the basal to the differentiated layers. A continuous regeneration throughout homeostasis and when skin injury occurs is maintained by the epidermal stem cells. Epithelial cells move upwards whereby the epithelial progenitor cells remain intact below the wound and the normal layer of epidermis will be recovered in around 3 days. If the basement membrane gets damaged, the normal epidermal cells from hair follicles, sweat glands and wound periphery will re- epithelialize the wound [3, 4, 24].
Epithelialization is the most essential part to immediately reconstruct skin barrier in wound healing where keratinocytes undergo a series of migration, proliferation and differentiation. Keratinocytes interact with fibroblasts, endothelial cells, immune cells, growth factors and cytokines in a balanced and coordinated manner in all phases of wound healing [3, 4, 25, 26].
Upon acute skin injury, neutrophils, monocytes, and macrophages are recruited to the injured site and after that keratinocytes become activated by the support of several cytokines and growth factors. Activation of keratinocytes is marked by the expression of K6 and K16, allowing keratinocytes to fill the defect in the wound.
Keratinocytes at the wound edge firstly must loosen their adhesion to completely heal the defect. Hemidesmosomes which link the basement membrane to the basal layer of keratinocytes need to be disassembled for cells to migrate [4, 27, 28].
This process is modulated and maintained through desmosomes and hemidesmosomes. PKCα is activated and converts calcium-independent to calcium-dependent desmosomes to lower their adhesive properties [29]. Transcription factor Slug allows additional keratinocytes release by increasing desmosomal disruption [30]. The α6β4 integrin which is expressed by basal keratinocytes binds to laminin-5 in the lamina densa of the basement membrane. It is hypothesized that there is a precursor form of laminin-5 which has a major binding affinity for α3β1, while the processed form has it’s binding affinity to α6β4 [31-34]. When keratinocytes migrate, α6β4 integrin switch to α3β1 integrin so laminin-5 binding occurs and K6, K16, and K17 keratins are upregulated. Serine phosphorylation of β4 subunits modulate α6β4 affinity for ligands through PKCα which increases keratinocyte motility by increasing disassembly of hemidesmosomes. EGF and macrophage-stimulating protein modulate PKCα- dependent phosphorylation. EGF and FGF stimulate keratinocyte migration and proliferation and induce the expression of K6 and K16 [35-37].
Cytokines play an important role in modulating migratory phenotypes of keratinocytes. IL-1 increases the secretion of FGF-7, IL-6 allows keratinocytes to respond to mitogenic factors and stimulate migration through the STAT3-dependent pathway. TGFβ1 promotes keratinocyte migration through MMPs (MMP-1, MMP-2, MMP-3, MMP-10, MMP-14, MMP-19, and MMP- 28) [38-43]. Keratinocytes also need to migrate through the fibrin and newly synthesized extracellular matrix of the wound for successful wound healing. MMP-1 maintains keratinocyte migration on type 1 collagen which is mediated by the α2β1 integrin, cytokines and growth factors. This process empowers keratinocytes to migrate from the wound edge, whereas keratinocytes behind the migrating tongue begin to proliferate [44-48].
During the advancement of migrating epithelial tongue, keratinocytes is the first layer that covers the wound, whichs start to proliferate, regulated by growth factors, cell differentiation rate, and attachment of cell to the substrate. Only the basal keratinocytes regain the ability to proliferate, while the terminal keratinocytes in the suprabasal layer don’t have this ability. Growth factors which are important in the proliferative process are HB-EGF, EGF, IGF-1, TGFα, and FGF-2. MMPs able to work together to assist growth factors in promoting keratinocyte proliferation through their proteolytic activity. They help to release growth factors from the wound matrix and can also digest the latent forms and convert them to active forms. The extracellular matrix is also able to engage integrins that will modulate growth factor receptor pathways. Foxn1 is known to be an important factor for basal keratinocytes proliferation, promotion of the initial differentiation, and transition from basal to suprabasal cells. Foxn1 participation in the epithelial mesenchymal transition indicates that epithelial mesenchymal transition process occurs in 4-6 days after skin injury which may be a critical time for scarless skin wound healing [49-56].
After the wound is fully epithelialized without drainage and covered by keratinocytes layer, the proliferation signaling pathway stops. Then, keratinocytes stratify to construct a cluster of cells in the superficial compartment. Behind the migrating epidermal tongue and toward the wound margin, keratinocytes stratify to renew progressively a differentiated epidermis which act as a barrier. Epidermal differentiation is balanced by the proliferation of keratinocytes where it only occurs in non differentiating layer and basal layer of the differentiating epidermis [57-59]
5. WOUND EPITHELIALIZATION MODELS
In order to evaluate keratinocyte migration in response to wounding there are several numbers of models using keratinocyte cultures, cultured or engineered human skin, and various animal models. Until now, although the researchers keep improving the existing models and developing a new one, there isn’t any model that can fully replicate human wound healing especially chronic wounds [60-63].
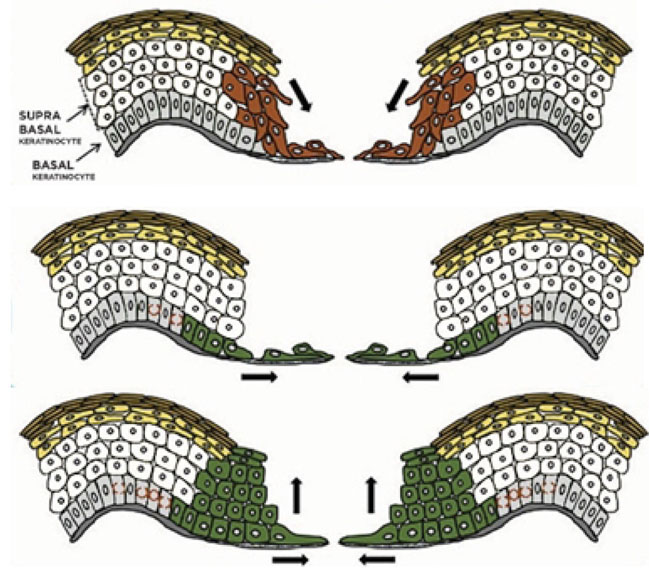
In this review, in general to all epithelialization models that have been proposed, we describe several mechanisms as shown in Fig. (2) that try to explain the recreation of epithelialization in wound healing [5, 24, 57].
The “tractor-tread” or “Sliding” theory explains that basal keratinocytes migrate over the wound bed and pull the remaining epidermis by expressing various integrins and maintain desmosomal junctions. Based on the Laplante et al. study, passive sliding of these superficial layers results from a pushing rather than from a dragging force which is shown by how the cornified layer had moved farther toward the wound center rather than the granular layer. It is suggested that the proliferating keratinocytes push the suprabasal cells toward the surface by mitotic pressure. Based on clonal lineage tracing, a model where the nonproliferating edge acts as a scaffold is shown. Most of the committed progenitor cells become highly proliferative and rapidly differentiate in the early wound healing phase. After that, basal cells increase the pool of stem cells later. This process prepares the wound bed for improved repopulation toward the centre and protects stem cells during wound healing [1, 5, 57, 64].
The ”leap-frogging” or “Rolling” theory explains that proliferating keratinocytes far from the wound edge enter from the suprabasal compartment and then are pushed onto the wound bed. After touching the wound bed, suprabasal keratinocytes would lower desmosome expression, then migrate over adherent basal keratinocytes, and after that change to basal phenotype. It is likely that the suprabasal cell slide over the leading basal cells to change to the basal cells. High expression of laminin-5 in the superficial layers during migration allows suprabasal keratinocytes located at the tip of the migrating epidermal tongue were elongated over to reach the connective tissue and the basal layer. Once they get in touch with the connective tissue, they soon become anchored and begin to deposit laminin 5 under the migrating epidermal tongue. These events allow an effective re-epithelialization in the wound center by the local keratinocytes proliferation and migration when the wound margin has become distant [1, 5, 57, 64].
Another variant of previous re-epithelialization model by evaluating keratins and Ki67 expression is a model by Usui et al. In this model, both basal and suprabasal cells activate as in both of the previous models (rolling and sliding) combined. Basal cells act as a leading tongue toward the center of the wound while suprabasal cells migrate, outnumbering the basal cells and becoming the primary source of cells. This model enables a large number of cells to quickly cover wound defects [64].
A three dimensional model for epidermal wound healing is presented by Safferling et al using integrated multiplex protein analysis, immunohistochemistry, and whole-slide imaging. During wound healing, the production of new keratinocytes forms of a concentric wave toward the centre of the wound. Hyperproliferation at the wound margins described as an initial positive signal after skin injury. The patterns of cell junction expression create a concentric field of basal keratinocytes which collectively migrates toward the wound. Basal cells collectively migrate from healthy area toward the wound. The suprabasal keratinocytes forms a shield with the cell junctions. This model showed that the extending epidermal tongue always migrates as a triangular multilayered epithelium with the full height toward the former wound margin and with a single cell tip at the protruding end toward the wound bed [24].
6. LESSONS FROM EPITHELIALIZATION MODEL
Impaired re-epithelialization might lead to chronic wound because it has failed to progress through the normal healing process and may enter a persistent inflammatory state. It can be caused by various pathological conditions such as diabetes, trauma and burns. In chronic wounds, the delay of re-epithelialization might be caused by bacterial infection, tissue hypoxia, exudates, local ischemia, and excessive inflammation. In this state, the cell pool is impaired and may demonstrate increased cellular senescence and a decreased cellular response to growth factors [65-69].
Excessive wound healing which is also caused by impaired epithelialization may result in the hypertrophic scars. Formation of scars produces serious cosmetic and functional implications. There is also a decrease in tensile strength compared to the normal skin. Previous studies stated that when wounds epithelialize in less than 14 days, almost no hypertrophic scar will be formed. Hypertrophic scar formation is caused by an unusual proliferation of fibroblasts and the deposition of excess ECM by the fibroblasts and myofibroblasts at the wound site. The keratinocytes in the epidermis of hypertrophic scars become active and produce growth factors that affect the inflammatory response, endothelial cells, and fibroblasts. All of these produce a cycle that keeps encouraging further formation of hypertrophic scars [70-73].
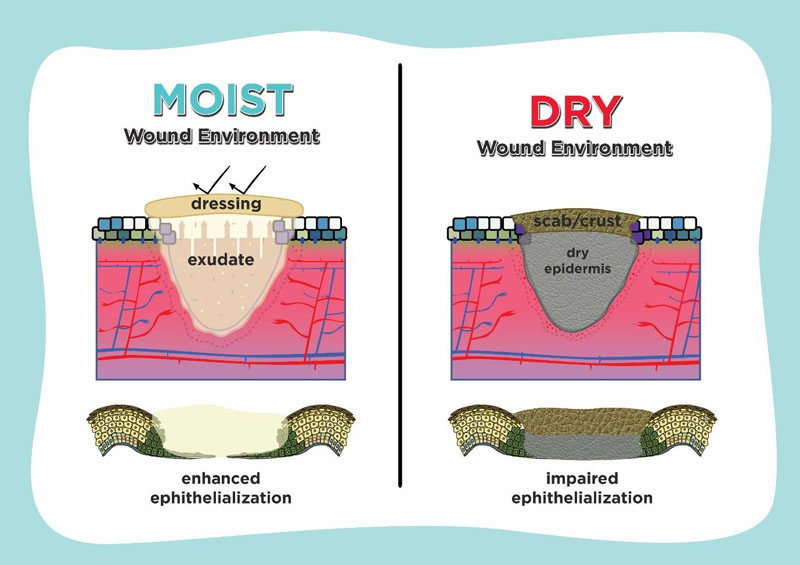
Understanding epithelialization promotes the idea of using moisture retentive occlusive dressings which enhance wound healing. If a dressing which controls hydration and holds moisture is used on the wound surface, it is defined as moist, whereas it is defined as dry if there isn’t any medium to contain the extracellular fluid and matrix. As shown in Fig. (3), in case of epithelialization, moist environment by using moisture-retentive dressing enhance epidermal cell migration especially in re-epithelialization. In contrast to the dry environment, cells usually dehydrate and die which cause scab or crust formation over the wound and impair re-epithelialization [7, 74, 75].
Enhanced wound healing in a moist environment is promoted by easier migration of epidermal cells on a moist surface, faster epithelialization, and prolonged presence of proteinases and growth factors. Several studies suggest that fluid in wounds healing promotes keratinocyte proliferation, fibroblast growth and preservation of growth factors. There was an acceleration of the inflammatory phase of repair in the moist wounds, so that the late phase of inflammation begins more rapidly [75, 76].
The physical barrier from the dry eschar tissue is an important factor for the delayed wound healing and poor epidermal migration in the dry environment. The delayed re- epithelialization observed in dry wounds on the fifth day after injury was associated with the presence of more wound debris than the moist wounds which appeared to impede epidermal cell migration. In the dry wound environment, scab formation limits the sliding of the superficial layers. Equilibrium between the proliferation, migration, stratification, and differentiation of keratinocytes in the wound should be maintained for an effective re-epithelialization [57, 76, 77].
Several studies have compared dry and moist wound environment for wound healing. Winter et al. showed that if the wound is exposed to the air and allowed to dry, it tends to heal slowly and result in poor cosmesis. Dyson et al. stated that moist wound environment improves inflammatory and proliferative phase in wound healing and enhances angiogenesis. Vogt et al showed that moist wound environment produces faster wound healing and less necrosis [75, 76].
Epithelialization model act as a basic mechanism that leads us to invent better products that aid in hard-healing wounds. Manufacturers from different countries produce multiple varieties of moist wound dressings such as hydrocolloids, foams, alginates and hydrogels. Wiechula et al in their systematic review showed that moist wound healing products have greater clinical advantage compared to non-moist products for managing split-thickness skin donor sites. Further studies are still needed to understand several unexplainable mechanisms in epithelialization. Through advanced understanding in this process, it would be possible that in the future, treatment of chronic wound can be further enhanced by newer products [74-77].
CONCLUSION
Epithelialization plays a major role in wound healing. Tthe epithelialization models give us an insight why moist wound environment is better than dry environment for treating wounds. No matter what the epithelialization model is, scab formation in dry environment will always impair wound healing. Because of that, treatment of wound in moist environment such as occlusive dressing is encouraged and will produce faster wound healing with less scar [57, 74-77].
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

